Computational Prediction of the Chromosome-Damaging Potential of Chemicals
Andreas Rothfuss,*,† Thomas Steger-Hartmann,† Nikolaus Heinrich,‡ and Jo¨rg Wichard‡,§
Experimental Toxicology, Schering AG, D-13342 Berlin, Germany, Computational Chemistry, Schering AG,D-13342 Berlin, Germany, and Molecular Modeling Group, FMP, D-13125 Berlin, Germany
We report on the generation of computer-based models for the prediction of the chromosome-damaging
potential of chemicals as assessed in the in Vitro chromosome aberration (CA) test. On the basis ofpublicly available CA-test results of more than 650 chemical substances, half of which are drug-likecompounds, we generated two different computational models. The first model was realized using the(Q)SAR tool MCASE. Results obtained with this model indicate a limited performance (53%) for theassessment of a chromosome-damaging potential (sensitivity), whereas CA-test negative compounds werecorrectly predicted with a specificity of 75%. The low sensitivity of this model might be explained bythe fact that the underlying 2D-structural descriptors only describe part of the molecular mechanismleading to the induction of chromosome aberrations, that is, direct drug-DNA interactions. The secondmodel was constructed with a more sophisticated machine learning approach and generated a classificationmodel based on 14 molecular descriptors, which were obtained after feature selection. The performanceof this model was superior to the MCASE model, primarily because of an improved sensitivity, suggestingthat the more complex molecular descriptors in combination with statistical learning approaches are bettersuited to model the complex nature of mechanisms leading to a positive effect in the CA-test. An analysisof misclassified pharmaceuticals by this model showed that a large part of the false-negative predictedcompounds were uniquely positive in the CA-test but lacked a genotoxic potential in other mutagenicitytests of the regulatory testing battery, suggesting that biologically nonsignificant mechanisms could beresponsible for the observed positive CA-test result. Since such mechanisms are not amenable to modelingapproaches it is suggested that a positive prediction made by the model reflects a biologically significantgenotoxic potential. An integration of the machine-learning model as a screening tool in early discoveryphases of drug development is proposed. Introduction
genotoxicity. Such screening strategies primarily rely on in Vitroassays, which often represent a cut down version of the re-
Screening approaches for determining the genotoxic potential
spective regulatory tests (e.g., Ames II) or make use of alter-
of new compounds play a pivotal role during hit validation and
native assays (e.g., the in vitro micronucleus test for the detec-
lead characterization phases of drug development in pharma-
tion of chromosomal damage). In principle, the concordance
ceutical companies. Traditionally, the assessment of the geno-
between screening assays and regulatory tests is relatively high
toxic potential of drug substances was typically performed
(2, 3). However, in particular with respect to screening assays
during early developmental stages by conducting a standard set
for chromosomal damage, they are at best medium throughput
(battery) of genotoxicity tests that support the submission of
and as such their use in early discovery stages is restricted
novel drugs to regulatory agencies. As outlined in the respective
because of costs and compound availability. Additionally,
ICH1 guidelines (1), this standard set generally consists of a
genotoxicity screens might be biased by the frequent presence
bacterial gene mutation test (Ames test), an in Vitro cytogenetic
of (genotoxic) impurities in early research drug batches leading
assay in mammalian cells for the detection of chromosomal
to potentially false positive results.
damage (e.g., a chromosome aberration (CA-) test) and an in
ViVo cytogenetic assay in rodent hematopoietic cells.
As an alternative, computational (in silico) structure-activity
Today, pre-regulatory genotoxicity tests are frequently per-
models have gained increasing importance in the assessment
formed in pharmaceutical companies because of increased
of a genotoxic potential. They have the clear advantage that no
compound throughput and in order to avoid late stage termina-
compound is needed for testing and that they can be applied in
tion of a cost-intensive drug development due to unforeseen
a true high-throughput manner. Computational programs usedfor genotoxicity prediction are mainly focusing on the prediction
* To whom correspondence should be addressed. Phone: +49-(0)30
of the outcome of the Ames test and relatively good predictive
46815268. Fax: +49-(0)30 46815091. E-mail: [email protected].
accuracies (>70%) can be reached for this endpoint (4). In
† Experimental Toxicology, Schering AG.
practice, however, it is not sufficient to solely predict bacterial
‡ Computational Chemistry, Schering AG. §
mutagenicity because results from in silico genotoxicity predic-
1 Abbreviations: CA-Test, chromosome aberration test; ICH, Interna-
tions are frequently used as part of the decision process during
tional Conference on Harmonisation of Technical Requirements for
drug discovery. Instead, it is desirable to also be able to model
Registration of Pharmaceuticals for Human Use; knn, k-nearest neighbour;
the chromosome-damaging potential of compounds in order to
QSAR, quantitative structure-activity relationship; SAR, structure-activity-relationship; SVM, support vector machine.
fully cover the basic regulatory mutagenicity tests. B Chem. Res. Toxicol.
However, in contrast to the Ames test prediction, no models
compounds tested positive in the CA-test, and thus, it seems
with comparable performance are currently available for the CA-
questionable as to whether similar performance characteristics
test. Several reasons might account for this situation. The good
and conclusions had been obtained using a more balanced data
correlation for the Ames test is based on the abundance of
set containing equal numbers of active and inactive compounds.
(publicly) available data for this test system as well as on the
Second, the structural diversity (chemical space) of compounds
fact that most of the molecular mechanisms underlying this
represented in the MCASE and machine learning model (12,
genetic endpoint are fairly well understood and can be directly
13) is clearly limited to mainly organic compounds, such as
related to the chemical structure (5). The situation is clearly
agrochemicals, known carcinogens, and industrial chemicals.
more complex for the CA-test. It is well-established that different
It was already noticed during the course of Ames-test modeling
mechanisms can lead to the microscopically visible formation
that computational models, which were predominantly con-
of aberrant chromosomes. Structural chromosome aberrations
structed using industrial and environmental compounds, per-
can be formed by direct drug-DNA interactions as a result of
formed in a clearly poorer manner when applied to pharma-
incorrect DNA repair processes (6) or an interaction of drugs
ceutical compounds (14-16). This is an important implication
with enzymes involved in DNA replication and transcription
if a computational prediction model for the CA-test has to be
(7). Numerical chromosome aberrations such as the gain or loss
developed as a screening tool during early drug discovery.
of chromosomes are generally a result of the interaction with
In the present study, we therefore aimed to construct and eval-
cellular proteins involved in chromosome segregation (8). In
uate two different computational models based on a heteroge-
addition, it is well-known that nonphysiological stimuli during
neous data set including a significant number of pharmaceutical
cell culture, such as those induced by excessive cytotoxicity,
compounds to be used in genotoxicity screening approaches in
osmolarity, pH and temperature, can also lead to structural
a pharmaceutical environment. The recent publication of two
data collections (10, 16) containing qualitative CA-test informa-
Furthermore, the CA-test is experimentally less standardized
tion on more than 650 compounds, including a significant
than the Ames test (i.e., different cells from different species
number of pharmaceuticals and drug-like compounds, allowed
are used), and publicly available experimental data is signifi-
us to readdress the issue of modeling a chromosome-damaging
cantly less abundant than Ames test data and almost purely
potential on the basis of the largest high-quality data collection
qualitative (i.e., aberration frequencies are hardly available).
Most importantly, the quality of available CA-test data isfrequently compromised by incomplete assay data sets and
Materials and Methods
differences in the judgment of a positive effect, in particular inthe presence of cytotoxicity (10). High-qualitative CA-test data
CA-Test Data Information. The CA-test data used in this study
might, in principle, be derived from publicly available data on
were obtained from two recently published data collections (10,
pharmaceuticals because they are likely to be conducted using
16). Further details on the original data source can be obtainedfrom the references of both data compilations.
ICH and GLP-compliant methods. However, such public data
The genotoxicity data collection from Snyder et al. (16) contains
are relatively scarce, and in particular, the number of positive
in Vitro cytogenetics data for 248 marketed pharmaceuticals, with
positive (i.e., chromosome-damaging) results being reported for
Consequently, only few publications are available in which
48/248 compounds (19%). Structural information could be retrieved
the performance of computational models for the prediction of
for 229 of the 248 compounds. Altogether, 189 negative and 40
CA-test data has been assessed. Using the MULTICASE
positive data records from this data source could be used for model-
(MCASE, Beachwood, USA) methodology for constructing
building purposes. As outlined in the article (16) and described in
experimental databases that can be used to predict the bioactivity
more detail in a previous collection effort (17), the in Vitro
of compounds, Rosenkranz et al. (11) reported the construction
cytogenetic data represents CA-test results obtained with diversecell types (Chinese hamster ovary cells, Chinese hamster lung cells,
of a CA-test prediction model based on 233 compounds. These,
V79 cells, MCL-5 human lymphoblastoid cells, and human blood
mostly organic compounds, were assessed in a CA-test as part
peripheral lymphocytes). Despite this obvious methodological
of the National Toxicology Program (NTP), with approximately
diversity, the overall quality of the data set and the reliability of
40% of the compounds being tested positive. Using an internal
the test result are judged to be high because the data has been
validation strategy, the observed sensitivity and specificity (i.e.,
generated according to standardized ICH- and GLP-compliant
the correct prediction of positives and negatives, respectively)
of the model were 53% and 71%, respectively (12).
The CGX database collected by Kirkland et al. (10) contains
More recently, Serra et al. (13) reported on the generation of
CA-test data for 488 structurally diverse compounds, consisting of
an automated machine-learning approach to generate classifica-
industrial, environmental, and pharmaceutical compounds. Out ofa total number of 488 chemicals, 292 (60%) were considered
tion models for the prediction of CA-test data. Support vector
positive, and 28 were judged to be equivocal. The latter were
machines (SVM) and k-nearest neighbor (knn) models were
excluded from our model building. Structural information was
developed on a set of molecular descriptors calculated for 346
retrieved for 450 out of the 460 remaining compounds. Altogether,
mostly organic compounds (29% positives). Using a prediction
168 negative and 282 positive data records from this data source
set of 37 compounds that were not included in model formation,
could be used for model-building purposes. Similar to the Snyder
sensitivity and specificity values of 73% and 92%, respectively,
CA-test collection, results obtained with all cell types are included
were obtained for knn classification models. Similar values were
in this compilation. With respect to data quality, considerable effort
was undertaken to review collected test results (10) suggesting anoverall consistent evaluation of test data. In order to estimate the
Despite the respectable performance characteristic, of the
number of drug-like compounds contained in this dataset, we
latter model in particular, their value for a routine in silico CA-
analyzed all 450 compounds for drug-likeness using a proprietary
test screening during early drug development seems to be
in-house software based on the model proposed by Sadowski and
questionable. First, the number of CA-test positive compounds
Kubinyi (18). Less than one-third of the compounds taken from
used for model building and evaluation in the Serra model (13)
Kirkland et al. (10) were considered as drug-like (data not shown),
appears to be critically low. Less than one-third of the
thus confirming that both data sources can roughly be separated
Modeling of Chromosome-Aberration-Test DataChem. Res. Toxicol. C Table 1. Data Sets Used for Model Generation Table 2. Performance Characteristics for the MCASE Model
Mean values of 10 independent validations. b Percentage of 2-8 atom
fragments structurally represented in the training set. Table 3. List of Some Significant Biophores Identified in the MCASE Model
average values for sensitivity (ratio of correctly predicted positive
into drug-like (16) and less drug-like (Kirkland et al., 2005)
compounds to all positives), specificity (ratio of correctly predicted
negative compounds to all negatives), and concordance (ratio of
As summarized in Table 1, 679 compounds were used in total
correctly predicted compounds to total number of compounds) were
for model generation, of which 322 tested positive (47%) and 357
tested negative (53%) in the CA-test. Machine Learning (ML) Model. For the machine-learning Collection of Structures. CAS numbers of identified substances
model, 10% of the data was randomly removed and used to assess
were collected from the respective data collections (10, 16) and
the performance of the final ML model (prediction set, see below).
queried in the MDL Toxicity database (MDL Information Systems
The remaining 90% of the data was designated as a training set
Inc., San Leandro, CA). The retrieved chemical structures were
stored as an sd file (MDL ISIS sdf file). For MCASE prediction
The process of ML model generation can be separated into three
model construction, SMILES notations of all compounds were
distinct processes. First, a broad set of molecular descriptors
generated by running the sd files through an existing prediction
encoding a variety of properties of the molecules are calculated
module in MCASE (Muticase Inc, Beachwood, OH), which
for each compound of the training set. Next, redundant information
generated a text file containing the respective SMILES code of the
of descriptors is removed via a process called feature selection,
resulting in a small subset of the most useful descriptors. Finally,
Model Construction and Validation in MCASE. A hallmark
a classification model is built on the basis of the identified
of the MCASE software is its capability to automatically generate
descriptors and validated using a set of data that was not previously
prediction modules on the basis of structural information and
included in the model-building effort.
associated bioactivity (19). Details on model generation and
Descriptor Generation and Feature Selection. All descriptors
software algorithms are published elsewhere (20). In essence, the
used in the ML model were calculated with the dragonX software
program identifies structural fragments, ranging from 2 to 10 atoms
(21) that was originally developed by Milano Chemometrics and
length, in combination with2D distances between atoms, which are
the QSAR Research Group. The software generates a total number
statistically correlated with activity (biophores) and inactivity
of 1664 molecular descriptors that are group into 20 different blocks,
(biophobes), respectively. In addition, the program detects fragments
such as constitutional descriptors, topological descriptors, and walk
that act as modulators of activity and takes into account basic
and path counts (22). For each compound in the training set, all
physicochemical descriptors for the module development process.
1664 descriptors were calculated. Because many of these descriptors
A limitation of MCASE is that compounds containing ions,
are redundant or carry correlated information, feature selection
molecular clusters (such as hydrates), and rare atoms (such as Mn,
processes need to be performed in order to select the most useful
Ca, or K) are not accepted for model generation. Consequently,
subset of descriptors to build a ML model.
compounds containing such structural features were automatically
Our feature selection approach follows the method of variable
eliminated from the training set by the program during model
importance as proposed by Breiman (23). The underlying idea is
to select descriptors on the basis of the decrease of classification
From the overall data set containing 679 data records, 100
accuracy after the permutation of the descriptors (24). Briefly, an
compounds (15%; 53 negative and 47 positive compounds) were
ensemble of decision trees is built, which uses all descriptors as
randomly removed before model building and used as a prediction
input variables and associated activity (CA-test result) as output
set to assess model predictivity. A training set was created out of
variables using 90% of the data (training set). The prediction
the remaining 579 compounds (304 negative and 275 positive
accuracy of the classification model on an out of training portion
compounds). Because of MCASE’s structural limitations, the
of the data (test set) is recorded. In a second step, the same is done
automatically generated MCASE model for CA-test prediction
after the successive permutation of each descriptor. The relative
contained 537 compounds (286 negatives and 251 positives). The
decrease of classification accuracy is the variable importance
predictivity of the generated model was assessed by internal and
following the idea that the most discriminative descriptors are the
external validation. For the internal validation, 10 separate, non-
most important ones. We first separately calculated the variable
overlapping sets consisting of 53 compounds (10% of the training
importance of each descriptor of the 20 blocks of molecular
set) were randomly selected from the training set and compiled as
descriptors and selected the most important ones. This descriptor
test sets. The remaining 90% of the individual learning sets were
set was reduced in a second iteration, resulting in a final set of 14
then used to predict the 53 compounds of the test set. For external
validation, the initially removed 100 compounds (prediction set)
Building the Machine Learning Classification Model. An
were predicted by the MCASE model. As performance parameters,
ensemble approach was used to build the final classification model
D Chem. Res. Toxicol. Table 4. List of DragonX Descriptors Used in the Machine-Learning
in model building. The procedure was independently repeated 20
times. This means that all model-building processes, that is, therandom removal of 10% of the data, the construction of a
classification model ensemble on the remaining 90% of the data
as outlined above (always using the same 14 dragon descriptors
determined in the feature selection step), and the prediction of both
training and prediction sets were performed each time. For a final
output, the mean average prediction values were calculated.
For the analysis of misclassified compounds, 50 independent
model-building rounds were performed, and the number of incorrect
classifications of each compound was recorded irrespective of its
presence in the training or test sets. Results and Discussion
On the basis of a data collection of high-quality CA-test
results of more than 650 pharmaceuticals and industrial chemi-
cals, we investigated the usefulness of two different computa-
tional approaches to predict the chromosome-damaging potential
of compounds. We used a functionality of the commercially
available MCASE system to automatically generate predictive
models from a training set of compounds with associated
qualitative (negative/positive) CA-test results. The predictivity
of an in-house prediction model built in MCASE and an in-
house machine-learning model in their ability to qualitatively
predict the outcome of the CA-test was assessed. MCASE Prediction Model. The performance characteristics
for the MCASE prediction model are listed in Table 2. Both
the training set and prediction set were predicted with compa-
rable performances. Sensitivity values for the training set andprediction set were 53% and 57%, respectively. Clearly, higher
for the prediction of the chromosome-damaging potential of the
values (i.e., 75% and 72%, respectively) were determined for
chemical compounds. An ensemble is the average output of several
the correct classification ratio of inactive compounds (specific-
different individual models, which were trained on different subsets
ity). Altogether, a concordance value of 65% was reached for
of the entire training data (sometimes called Bootstrap Aggregating
or Bagging, (25)). Building ensembles is a common way to improve
Interestingly, the performance characteristics obtained in our
classification and regression models in terms of stability and
study were very similar to those reported by Rosenkranz et al.
accuracy. We built heterogeneous ensembles consisting of severaldifferent model classes to achieve diverse ensembles (26). The
(12), although their data set was much smaller in size (n ) 233
model classes were as follows: (1) classification and regression
vs n ) 537 in our study). The Danish-EPA reports on their
trees (CART), where we used the implementation in the MATLAB
website (http://www.mst.dk/) on the creation of an MCASE
Statistics Toolbox (The MathWorks, Natick, USA); (2) support
model based on approximately 500 chromosome aberration test
vector machines (SVM) with Gaussian kernels (28); (3) linear
data taken from the Ishidate data collection (31). Although
discriminant analysis (LDA), quadratic discriminant analysis (QDA),
overall higher performance values for this model were reported
and linear ridge models (29); (4) feedforward neural networks (NN)
(76% concordance), similar unbalanced values for sensitivity
with two hidden layers trained with a simple gradient descend (30);
(59%) and specificity (82%) were achieved. The persistently
and (5) k-nearest-neighbor models (knn) with adaptive metrics (30).
low sensitivity of the MCASE models indicates that the
The selection of the different model classes used for the
underlying 2D-fragment-based descriptors do not sufficiently
construction of the final classification model was based on cross-
describe the mechanism(s) leading to a positive result in the
validation (CV) approaches. This means that the training set (i.e.,
chromosome-aberration test. One way to further assess this
90% of the total data) was split randomly into a training-learning
possibility is the analysis of identified structural fragments that
set (80% of the data) and a training test set (20% of the data). Each of the different model classes was then trained on the training-
are statistically correlated with activity (biophores).
learning set and assessed for their prediction accuracy on the training
A list of the most significant biophores identified in our
test set. This procedure was repeated 21 times using a novel
MCASE model is given in Table 3. As can be seen from the
randomly selected training-learning and training test set each time.
respective structural representation, almost all identified bio-
In each of the runs, only the best model (i.e., the one showing the
phores represent known structural alerts for DNA reactivity. This
lowest classification error) was selected to become a member of
implies that the structural determinants that on the basis of our
the final ensemble. In this way, all model classes had to compete
MCASE analysis contribute to a positive effect in the chromo-
with each other because they are trained and tested on the same
some aberration test reflect a direct drug-DNA (i.e., electro-
data set. Our approach, thus, resulted in a final classification model
philicity) interaction and, thus, are identical to the structural
(the ML model) consisting of 21 individual models. The predictionoutput of this ML model is based on the counting of the vote of
fragments identified from Ames test data (32).
each of the individual 21 models and the determination of the
The low sensitivity of the MCASE prediction model clearly
majority vote, which then constitutes the final prediction.
limits its application as a decision tool during lead characteriza-
Performance Evaluation of the ML Model. In a final step,
tion phases. Companies developing new compounds are pri-
the performance of the ML model was assessed on the entire
marily dependent on prediction tools that have a relatively low
training set (90% of the total data) and on the 10% of data
false negative prediction rate (i.e., high sensitivity) in order to
(prediction set), which was initially removed and never included
focus further development on those compounds that are presum-
Modeling of Chromosome-Aberration-Test DataChem. Res. Toxicol. E Table 5. Performance Characteristics for the Machine-Learning Model a TP, true positive; FN, false negative; TN, true negative; FP, false positive. b Values represent mean ( SD of 20 independent validations. Table 6. Performance Comparison between the Present ML model
ably safe. However, false positive predictions could result in
and the knn and SVM models published by Serra et al. (13)
the loss of valuable candidates. Therefore, a balanced perfor-mance between sensitivity and specificity is desirable, resulting
in ideal predictive tools that show equally high values for
sensitivity, specificity, and concordance. This, however, is
ML modelb
clearly not the case for the MCASE model, where the acceptableconcordance value is primarily based on the low false positive
a Values taken from Serra et al. (13). b Only part of the Serra dataset
rate. In other words, the particular descriptor applied in MCASE
was used for the analysis. For further details, see the Results and Discussionsection.
seems to be limited to pick up only one mechanism of CAinduction, that is, the direct interaction of a drug with DNA. In
vs 75% for the training set), resulting in a balanced prediction
order to overcome this apparent limitation, we investigated
model with almost equal performance values for sensitivity,
whether the use of more complex molecular descriptors in
combination with a machine-learning approach might enable
Although this improvement reflects the usefulness of applying
us to generate more predictive classification models.
various molecular features as discriminators for the prediction
Machine Learning Model. Statistical learning methods, such
of chromosome-damaging potential, a comparison with the
as support vector machines (SVM) or k-nearest-neighbor (knn)
values reported by Serra et al. (13) might lead to the conclusion
approaches are currently being used as a new approach in in
that our ML model has a lower performance. However, a direct
silico toxicity prediction (33, 34). Compared to traditional QSAR
comparison of performance values between this study and Serra
modeling approaches, statistical learning methods are often
et al. (13) is difficult because of the differences in the data set
superior in terms of performance (35). As outlined in detail in
and statistical evaluations. As mentioned before, Serra et al.
the Materials and Methods section, we used a novel approach
used a smaller (and structurally less diverse) prediction set in
by building a classification model based on a heterogeneous
which the proportion of known chromosome-damaging com-
ensemble of SVM, knn, neuronal networks, and other model
pounds was lower than that in our study (11 out of 37
compounds vs 70 out of 145 in our study). Thus, it remains
A list of the 14 molecular descriptors selected for model
open as to whether similarly good performance values would
building purposes are given in Table 4. As outlined in detail in
be achieved if a more extensive prediction set containing more
the Materials and Methods section, these 14 descriptors were
CA-test positive compounds had been used. Second, the model
selected from more than 1600 dragonX descriptors after
characteristics described by Serra et al. seem to be based on a
eliminating those that are redundant and choosing those that
single cross-validation effort only, whereas we used a 20-fold
had the highest impact for classification. Several of the identified
CV to perform our validation procedure.
descriptors can be directly related to genotoxicity and, thus,
Despite these differences in model construction, we attempted
present a mechanistically sound basis of the molecular features.
to get a more objective comparison of the predictive value of
Several functional descriptors as well as (electro)topological
our ML model by applying it to the compound data used by
indices specify characteristics of structures involved in DNA
Serra et al. (13). In order to not be biased by our training set,
modifications. Generic descriptors, such as geometrical and
only those compounds that were not included in our training
general information indices, describe the shape, size, and
set were extracted and, thus, represent novel compounds.
composition of molecules. A recent study on the prediction of
Altogether, 291 compounds fulfilled this criterion, out of which
genotoxicity by using statistical methods, such as SVMs and
74 were reported with a positive result. These compounds were
knn, indicates that such generic descriptors can be valuable for
then collected as sd files, computed with our set of 14 molecular
describing the DNA-reactive property of compounds (33).
descriptors. and classified using our ML model. The resulting
Molecular weight was selected as a discriminating feature
performance characteristics are given in Table 6 in comparison
probably because of the heterogeneous data base, consisting of
to the values reported by Serra et al. (13). Because the selected
many small organic chemicals that are chromosome-damaging
compounds were not previously included in our ML model, they
(10) and an equally large amount of pharmaceutical compounds
can be seen as an independent prediction set, which we
that are mostly not chromosome-damaging (16).
compared to our data. Overall, this comparison shows that our
The performance values of our machine-learning model for
ML model reaches comparable prediction accuracies to those
the training set and prediction set are given in Table 5. As
of the learning models reposted by Serra et al., although the
outlined in Materials and Methods, 20 independent cross
latter were trained on a structurally less diverse set of com-
validations were performed by removing each time 10% of the
pounds. Nevertheless, the sensitivity of our ML model clearly
data (prediction set), building the ML model using the remaining
outscores the performance characteristics of the knn and SVM
90% of the data (training set), and then predicting the removed
compounds. The values for true positive, false negative, true
Although tentative in nature, several conclusions can be drawn
negative, and false positive predictions of both training and
from this comparison. First, it is reasonable to assume that the
prediction sets as well as for the other performance character-
lower prediction accuracies observed with our test set data
istics outlined in Table 5, thus, represent the mean ( standard
compared to that of Serra et al. (13) is a consequence of the
deviation of 20 independent evaluations. Compared to the data
extension of the chemical space in our training set by adding a
obtained with the MCASE model, the ML approach led to a
significant amount of pharmaceutical compounds to the less
clearly improved prediction of CA-positive compounds (53%
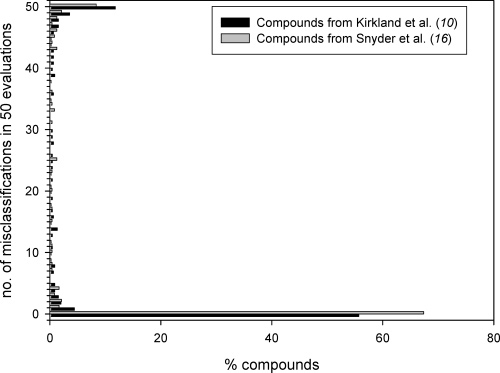
drug-like compounds contained in the Kirkland data set (10). F Chem. Res. Toxicol. Table 7. List of 15 False Negative Classified Pharmaceuticals Figure 1. Percentage of compounds from both data sources (Kirkland
et al., (10); Snyder et al. (16)) plotted against the number of incorrect
predictions (misclassification) in a series of 50 independent evaluations.
A compound that was correctly predicted in all of the runs, thus, falls
into the group of zero misclassifications, whereas a consistently
incorrect predicted compound is classified into the group of 50
a Genotoxicity information taken from Snyder et al. (10). b Ames, Ames
test; BM, mouse bone marrow micronucleus test; N/A: not available.
Because the majority of CA-test positive compounds in ourstudy originates from the Kirkland data compilation, which from
in only five cases (not listed). Mechanistic information on a
a chemical diversity point of view resembles the Serra data, it
possible mode of action of chromosome-damage induction of
is not surprising that our ML model performs particularly well
the 15 known genotoxic compounds is limited. Most of the
in terms of sensitivity on the latter data set. The development
compounds do not contain structural alerts for mutagenicity,
of prediction models for diverse data sets, such as those in our
suggesting that they do not primarily act genotoxic through
study, is generally considered to be problematic (36), and in
direct drug-DNA interaction. A review of other mutagenicity
theory, the construction of two local models (i.e., one for each
test results obtained for the false-negative-predicted compounds
data set) would have been favorable. Such an approach,
shows 5 out of 14 compounds (no mutagenicity data were
however, is currently not feasible, because only few CA-test
available for imipramine) were also tested positive in an Ames
positive data for drug-like compounds are publicly available,
test, suggesting a genotoxic potential that was missed by our
and sufficiently large training sets for CA-test modeling,
ML model. Surprisingly, 9 out of the 14 compounds were tested
therefore, need to be compiled from structurally diverse
positive uniquely in the CA-test, whereas they yielded negative
compounds, as has been done in our study.
results in the Ames-test and the in vivo mouse micronucleus
Given the structural diversity of our training set used for
test (Table 7). This suggests that the positive CA-test result of
model construction, we investigated whether our ML model
these misclassified compounds might not be due to an inherent
performed differently on the two underlying data sets. As a
genotoxic potential but instead induced by biologically non-
measure for predicting accuracy, we determined the number of
significant effects detected by this test system.
misclassifications for each compound of both data sources in
As outlined before, nonphysiological stimuli during cell
50 independent evaluations. This means that an ML model was
culture can lead to structural chromosome aberrations (9). It is
generated 50 times, and in each run, the classification result
likely that other yet unknown mechanisms that are not directly
(i.e., true or false) was recorded for each compound. A
related to the chemical structure can result in a (biologically
compound that was correctly predicted in all of the 50 runs
not significant) positive result in the CA-test. Because these
would, thus, be categorized with zero misclassifications, whereas
artificial effects are not directly related to the chemical structure
a compound showing 50 misclassifications would have always
of the compound, they are not amenable to modeling and,
been predicted incorrectly. The results of this exercise are shown
therefore, automatically decrease the predictivity of computa-
in Figure 1. As can be seen, almost 70% of all compounds from
the pharmaceutical class (16) were correctly predicted in 50 out
In conclusion, our data show that the chromosome-damaging
of 50 evaluations (i.e., zero misclassifications). In comparison,
potential of pharmaceuticals can be predicted using machine-
the same was true for less than 60% of the less drug-like class
learning approaches, albeit with lower predictivity than that
(10). However, approximately 10% of pharmaceuticals were
previously reported for industrial chemicals (13). Nevertheless,
never predicted correctly (50 misclassifications), which to a
the inclusion of a significant amount of pharmaceutical com-
slightly higher degree was also true for the Kirkland compounds.
pounds into our model and the concomitant expansion of the
Altogether, it can be stated that compounds from the pharma-
chemical space covered by the model now makes it a potentially
ceutical class were predicted with higher accuracies than those
useful tool that can be incorporated in compound selection
processes during early phases of drug development. A balanced
Of the 20 compounds from the pharmaceutical class that were
prediction accuracy of 70-75% is sufficiently high during these
consistently misclassified in all 50 evaluations, 15 are false
developmental phases to filter out potential genotoxic com-
negatives, that is, a chromosome-damaging potential was missed.
pounds. Together with an experimental screening test (e.g., the
These 15 false negatives are listed in Table 7. Compounds were
in Vitro micronucleus test) for the follow-up testing of com-
incorrectly classified as chromosome-damaging (false positives)
pounds with a negative call, such a tool can significantly
Modeling of Chromosome-Aberration-Test DataChem. Res. Toxicol. G
contribute to a more targeted development of non-genotoxic drug
(16) Snyder, R. D., Pearl, G. S., Mandakas, G., Choy, W. N., Goodsaid,
candidates. In addition, given the high concordance between
F., and Rosenblum, I. Y. (2004) Assessment of the sensitivity of thecomputational programs DEREK, TOPKAT and MCASE in the
the in Vitro micronucleus test and the CA-test, data obtained
prediction of the genotoxicity of pharmaceutical molecules. EnViron.
during the experimental screening of drug compounds could
be fed back in order to train improved models solely based on
(17) Snyder, R. D., and Green, J. W. (2001) A review of the genotoxicity
of marketed pharmaceuticals. Mutat. Res. 488, 151-169.
(18) Sadowski, J., and Kubinyi, H. (1998) A scoring scheme for discrimi-
nating between drugs and nondrugs. J. Med. Chem. 41, 3325-3329. References
(19) Klopman, G., and Rosenkranz, H. S. (1994) International Commission
for Protection Against Environmental Mutagens and Carcinogens.
(1) ICH 2SB: Genotoxicity: a standard battery for genotoxicity testing
Approaches to SAR in carcinogenesis and mutagenesis. Prediction of
for pharmaceuticals. CPMP/ICH/174/95.
carcinogenicity/mutagenicity using MULTI-CASE. Mutat. Res. 305,
(2) Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D.,
and Madle, S. (1998) Evaluation of the in vitro micronucleus test as
(20) Rosenkranz, H. S., Cunningham, A. R., Zhang, Y. P., Claycamp, H.
an alternative to the in vitro chromosomal aberration assay: position
G., Macina, O.T., Sussmanm, N. B., Grant, G. S., and Klopman, G.
of the GUM working group on the in vitro micronucleus test. Mutat.
(1999) Development, characterization and application of predictive
toxicology models. SAR QSAR EnViron. Res. 10, 277-298.
(3) Diehl, M. S., Willaby, S. L., and Snyder, R. D. (2000) Comparison
(21) http://www.talete.mi.it/dragon_exp.htm
of the results of a modified miniscreen and the standard bacterial
(22) Todeschini, R., and Consonni V. (2000) Handbook of Molecular
reverse mutation assays. EnViron. Mol. Mutagen. 35, 72-77.
Descriptors. In Series of Methods and Principles in Medicinal
(4) White, A. C., Mueller, R. A., Gallavan, R. H., Aaron, S., and Wilson,
Chemistry, (Mannhold, R., Kubinyi, H., and Timmerman, H., Eds.)
A. G. (2003) A multiple in silico program approach for the prediction
Vol. 11, Wiley-VCH, Weinheim, Germany.
of mutagenicity from chemical structure. Mutat. Res. 539, 77-89.
(5) Simon-Hettich, B., Rothfuss, A., and Steger-Hartmann, T. (2006) Use
(23) Breiman, L. (2001) Random forests. Machine Learning 45, 5-32.
of computer-assisted prediction of toxic effects of chemical substances.
(24) Breiman, L. (1998) Arcing classifiers. Annals of Statistics 26, 801-
(6) Obe, G., Pfeiffer, P., Savage, J. R., Johannes, C., Goedecke, W.,
(25) Breiman, L. (1996) Bagging predictors. Machine Learning 24, 123-
Jeppesen, P., Natarajan, A. T., Martinez-Lopez, W., Folle, G. A., and
Drets, M. E. (2002) Chromosomal aberrations: formation, identifica-
(26) Wichard, J., and Ogorzalek, M. (2006) Time series prediction with
tion and distribution. Mutat. Res. 504 17-36.
ensemble models applied to the cats benchmark. Neurocomputing, in
(7) Degrassi, F., Fiore, M., and Palitti, F. (2004) Chromosomal aberrations
and genomic instability induced by topoisomerase-targeted antitumour
(27) Breiman, L. (1993) Classification and Regression Trees. Chapman &
drugs. Curr. Med. Chem.: Anti-Cancer Agents 4, 317-25.
(8) Parry, E. M., Parry, J. M., Corso, C., Doherty, A., Haddad, F., Hermine,
(28) Chang, C., and Lin, C. (2001) Libsvm - A library for support vector
T. F., Johnson, G., Kayani, M., Quick, E., Warr, T., and Williamson,
machines. http://www.csie.ntu.edu.tw/∼cjlin/libsvm.
J. (2002) Detection and characterization of mechanisms of action of
(29) Hastie, T., Tibshirani, R., and Friedman, T. (2001) The Elements of
aneugenic chemicals. Mutagenesis 17, 509-21.
Statistical Learning. In Springer Series in Statistics (Bickel, P., Diggle,
(9) Kirkland, D., and Mu¨ller, L. (2000) Interpretation of the biological
P., Fienberg, S., Gather, U., Olkin, I., and Zeger, S., Eds.) Springer-
relevance of genotoxicity test results: the importance of thresholds.
(30) Merkwirth, C., and Wichard, J. (2002) ENTOOL - A MATLAB
(10) Kirkland, D., Aardema, M., Henderson, L., and Mu¨ller, L. (2005)
toolbox for ensemble modelling, http://chopin.zet.agh.edu.pl/∼wichtel/.
Evaluation of the ability of a battery of three in vitro genotoxicitytests to discriminate rodent carcinogens and non-carcinogens. I.
(31) Sofuni, T. Ed. (1998) Data Book of Chromosomal Aberration Test in
Sensitivity, specificity and relative predictivity. Mutat. Res. 584,
Vitro. Life Science Information Center, Japan.
(32) Ashby, J., and Styles, J. A. (1978) Does carcinogenic potency correlate
(11) Rosenkranz, H. S., Ennever, F. K., Dimayuga, M., and Klopman, G.
with mutagenic potency in the Ames assay? Nature 271, 452-455.
(1990) Significant differences in the structural basis of the induction
(33) Li, H., Ung, C. Y., Yap, C. W., Xue, Y., Li, Z. R., Cao, Z. W., and
of sister chromatid exchanges and chromosomal aberrations in Chinese
Chen, Y. Z. (2005) Prediction of genotoxicity of chemical compounds
hamster ovary cells. EnViron. Mol. Mutagen. 16, 149-177.
by statistical learning methods. Chem. Res. Toxicol. 18, 1071-1080.
(12) Rosenkranz, H. S. (2004) SAR modelling of genotoxic phenomena:
(34) Zhao, C. Y., Zhank, H. X., Zhang, X. Y., Liu, M. C., Hu, Z. D., and
the consequence on predictive performance of deviation from a unity
Fan, B. T. (2006) Application of support vector machine (SVM) for
ratio of genotoxicants/non-genotoxicants. Mutat. Res. 559, 67-71.
prediction toxic activity of different data sets. Toxicology 217, 105-
(13) Serra, J. R., Thompson, E. D., and Jurs, P. C. (2003) Development of
binary classification of structural chromosome aberrations for a diverse
(35) He, L., Jurs, P. C., Custer, L., Durham, S. K., and Pearl, G. M. (2003)
set of organic compounds from molecular structure. Chem. Res.
Predicting the genotoxicity of aromatic compounds from molecular
structure with different classifiers. Chem. Res. Toxicol. 16, 1576-
(14) Cariello, N. F., Wilson, J. D., Britt, B. H., Wedd, D. J., Burlinson, B.,
and Gombar, V. (2002) Comparison of computer programs DEREK
(36) Richard, A. M., and Benigni, R. (2001) AI and SAR approaches for
and MCASE to predict bacterial mutagenicity. Mutagenesis 17, 321-
predicting chemical carcinogenicity: survey and status report. SARQSAR EnViron. Res. 13, 1-19.
(15) Greene, N. (2002) Computer systems for the prediction of toxicity:
an update. AdV. Drug DeliVery ReV. 54, 417-431.
Dr. rer. nat. Frank GollnickForschungsgemeinschaft FunkJuutilainen, 1997: Juutilainen J, Lang S: “Genotoxic, carcinogenic and teratogenic effects of electromagnetic fields. Introduction and overview” in: Mutat Res 1997; 387 (3): 165 - 171Brusick, 1998: Brusick D, Albertini R, Mc Ree D, Peterson D, Williams G, Hanawalt P, Preston J: “Genotoxicity of radiofrequency radiation. DNA/Genetox Exp
Jahreshauptversammlung -Wie bei der Vorstandssitzung vor dem Perseiden-Feuer beschlossen, wird die Mitgliederversammlung erst am Nationalfeiertag und nicht bei der Linzer Klangwolke stattfinden, da die Vorarbeiten angesichts erheblicher Überstunden unseres Mitgliederbetreuers noch nicht weit gediehen sind. Zeitpunkt ist 17 Uhr, der genaue Ort wird erst nach Rücksprache mit den Projektgruppe
 F Chem. Res. Toxicol.
F Chem. Res. Toxicol.